

In 1984, the Human Genome Project (HGP) utilized First Generation Sequencing methods, specifically Sanger Sequencing, to complete the sequencing of the entire human genome, consisting of 3 billion base pairs. This monumental endeavor took over a decade and cost approximately 3 billion dollars.
With the evolution and development of sequencing technologies, the advent of Next Generation Sequencing (NGS) in 2007 significantly reduced the cost and time required for sequencing. Also known as Second Generation Sequencing or High-Throughput Analysis, this efficient sequencing technology now boasts single-run outputs of up to 540-600 Gb (Illumina system). Coupled with advanced bioinformatics analysis techniques, NGS has been widely applied across various academic disciplines to study genomics, transcriptomics, and epigenomics.
Third Generation Sequencing (also known as Long-Read Sequencing) is a sequencing technology where the average sequence read length can exceed 10,000 base pairs (10 kb). This technology enables the direct sequencing of raw DNA samples, eliminating the need for the PCR amplification process required by Second Generation Sequencing technologies. This approach bypasses the error rates and biases associated with PCR amplification and allows for the analysis of DNA methylation modifications.
Prominent technologies in the Third Generation Sequencing field include "Single Molecule Real-Time Sequencing" (SMRT) introduced by Pacific BioSciences (PacBio) in 2010 and the "Nanopore-Based Platform" launched by Oxford Nanopore Technologies (ONT) at the end of 2012. The long sequencing fragments provided by Third Generation Sequencing systems have found applications not only in genomics, transcriptomics, and epigenomics research but also in a broader range of fields such as biomedical and biotechnological research.
Compared to traditional First and Second Generation Sequencing, Third Generation Sequencing has made significant progress in improving speed, reducing costs, and enhancing data quality. This progress provides researchers with more choices to meet their specific needs. Notable platforms utilizing Nanopore technology include Flongle, MinION, GridION, and the latest PromethION 2 Solo, launched at the end of 2022.
The Advantages of Tri-I Next Generation Sequencing Services :
1. Nanopore and Illumina offer a complete range of sequencing platforms for long and short sequences.
2. An excellent choice for high efficiency and quality.
3. Professional sample quality control processes and library preparation.
4. Professional bioinformatics analysis and service consultation.
- To learn more about issues related to Next Generation Sequencing Services, please click the NGS FAQs.
- If you would like to entrust Next Generation Sequencing Services, please click on the Laboratory Login. After logging in, you can proceed with the order.
- If you need to download the sample testing service order form for Next Generation Sequencing, please click on the DOWNLOAD button.
Illumina

Next Generation Sequencing (NGS)
Next Generation Sequencing (NGS) also known as next-gen sequencing or high-throughput sequencing, is a new technology developed based on the first-generation sequencing methods. The limitations of the first generation methods, such as low throughput, high costs, and time-consuming procedures, posed challenges for large-scale applications, leading to the development of NGS technology. NGS significantly reduces the cost per base sequenced, eliminating the limitations of gene size or abundance in current sequencing methods. It involves fragmenting sequences to attach sequencing adapters to create sequence libraries, which are then processed through NGS sequencers, enabling molecular amplification. High-throughput sequencing techniques can be applied across diverse genomic studies using various library construction methodologies, including applications such as Targeting Gene Sequencing, RNA Sequencing, and Metagenomics.
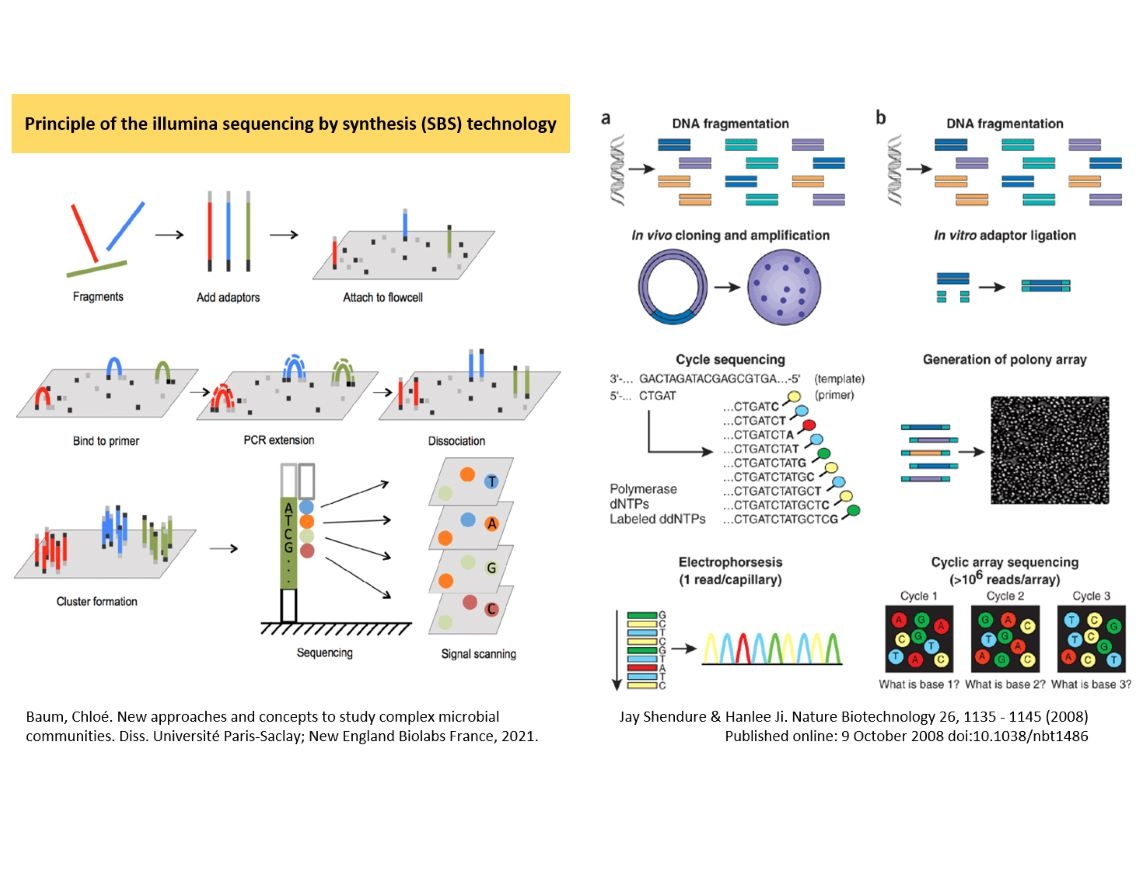
Currently, Illumina's next generation sequencers dominate the market. The image illustrates a brief overview of Illumina sequencing principles.
Nanopore

Third Generation Sequencing (Nanopore)
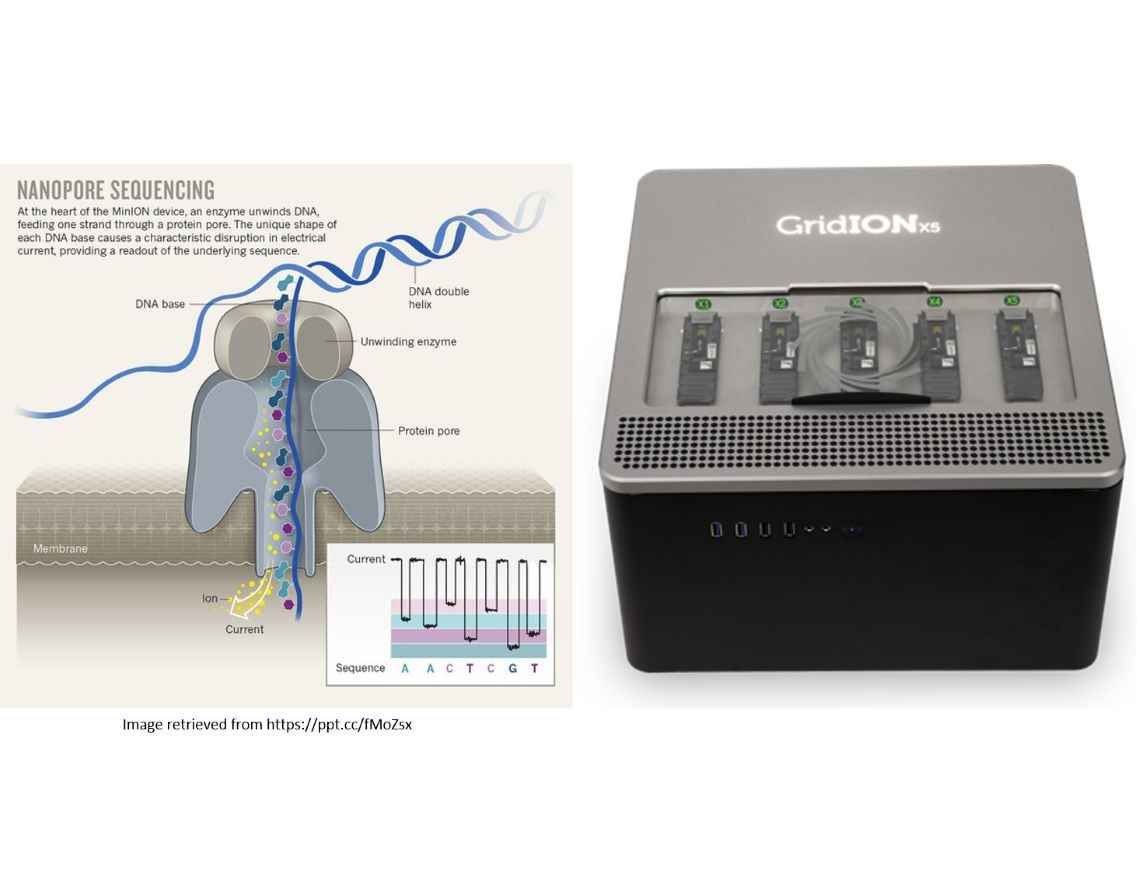
Nanopore sequencing, a third generation sequencing technology for nucleic acids (RNA and DNA), uses physical methods to capture molecules within specialized nanopores via covalent bonds. As individual DNA or RNA bases pass through these nanopores, they induce changes in electric charge, briefly altering the current strength. Real-time monitoring of these fluctuations helps identify the specific bases being passed. This technology differs from previous gene sequencing methods as it doesn't require polymerase chain reactions or chemical labeling of samples; instead, it sequences individual DNA or RNA molecules directly and instantly, allowing for real-time analysis of DNA or RNA segments of any length. Moreover, it can produce single-read sequences exceeding 1 Mb, providing significant support for genome assembly. Nanopore sequencing offers cost-effective individual genotyping, high portability, simplified and rapid sample processing, and immediate analysis of results.
Currently, nanopore sequencing finds widespread application in various domains, including human genome and methylation sequencing, genomics of animals and plants, pathogen genome sequencing for rapid diagnostics, environmental or food microbial monitoring, and numerous other related applications.
Whole Genome Sequencing
Whole Genome Sequencing (Illumina)
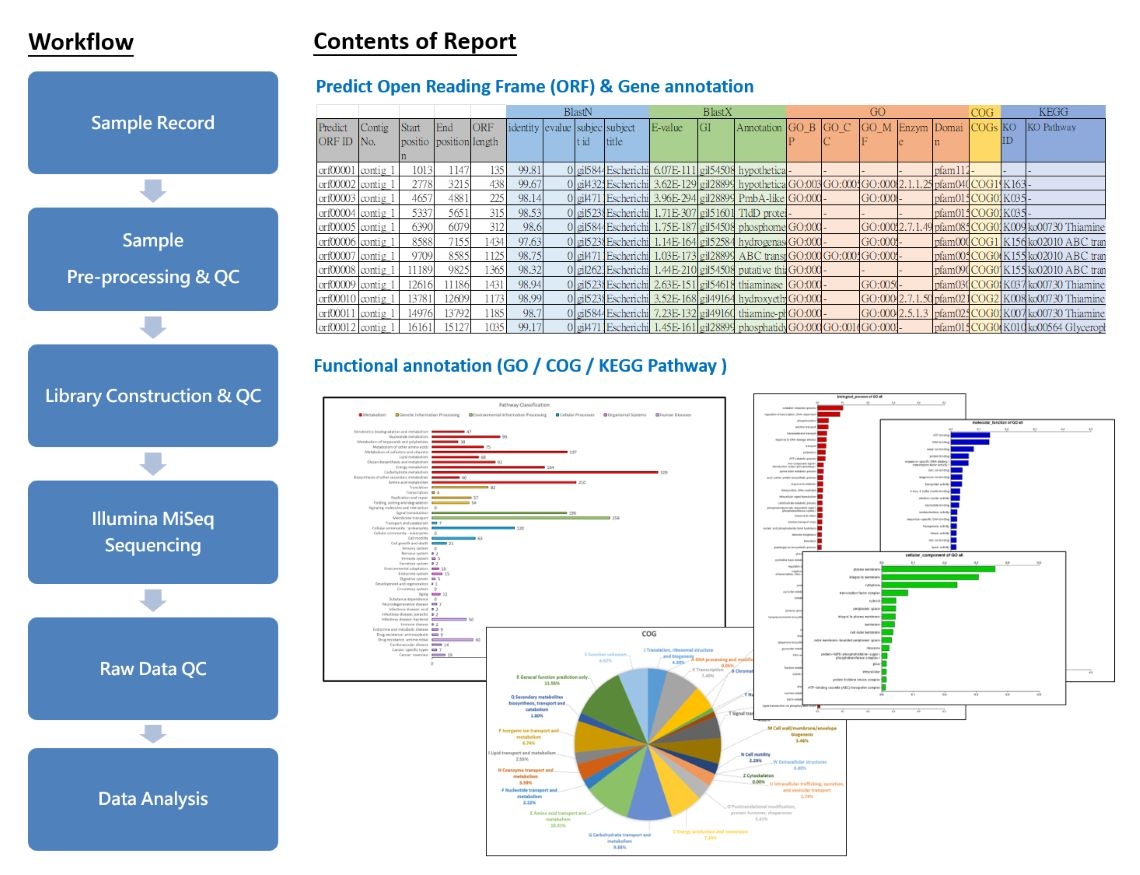
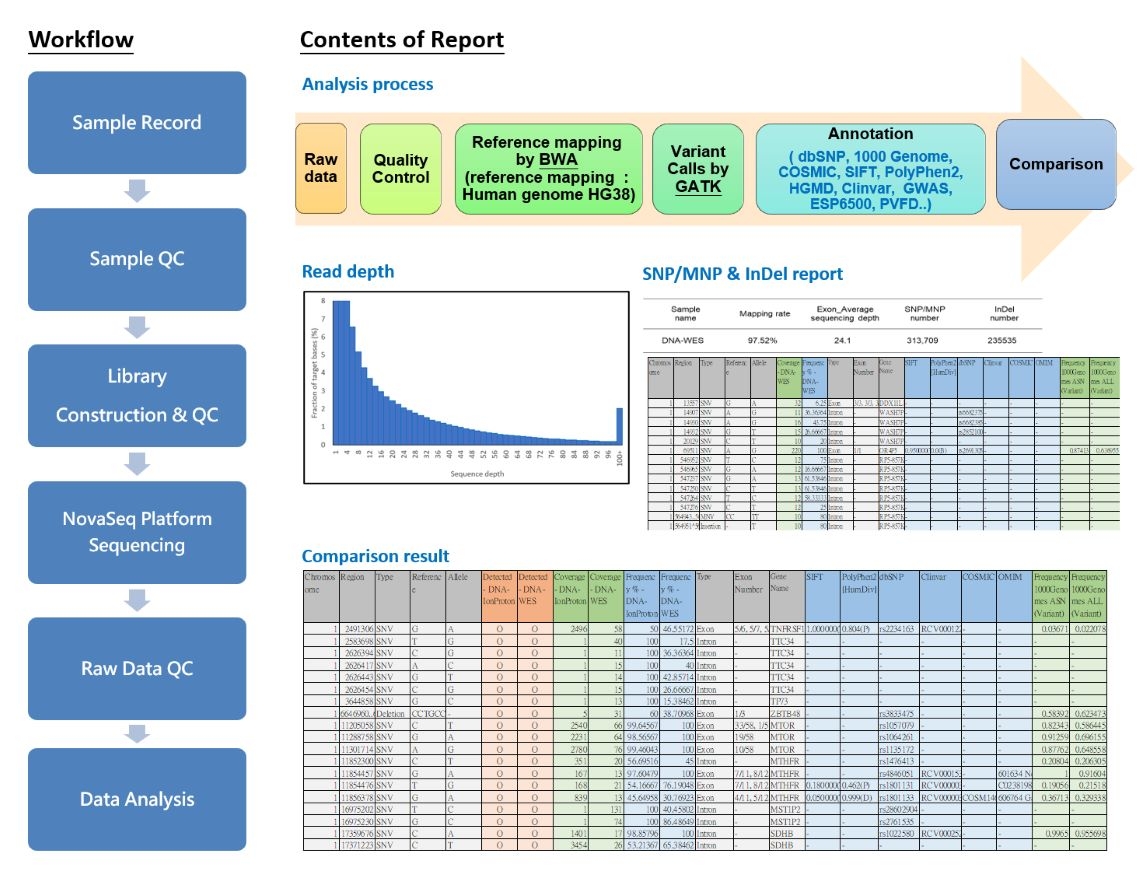
Through the next generation sequencing system Illumina, the DNA sequences of individuals, whether known or unknown, can be sequenced to obtain information on gene sequences, genetic variations, gene functionality, and important insights for drug research. This method allows for differential analysis at the individual or population level to explore disease-related issues.
Currently, there are two Illumina platforms available for data acquisition: MiSeq (PE300) and NovaSeq (PE150). During whole-genome sequencing, samples are fragmented to sizes suitable for sequencing, followed by library construction and sequencing on the selected platform. The reports include quality control (QC) of samples and data, assembly and alignment of sequences (BlastN, BlastX), database analysis (GO, COG, KEGG), and Genome map, as well as customized analysis services.
Sample Requirements :
Sample Purity: OD 260/280 ratio should be between 1.7 and 1.9.
Sample Concentration: Minimum concentration should not be less than 100 ng/µl.
Total Sample Amount: The recommended total amount is 5 µg, with a minimum of at least 1 µg.
Sample Solvent: Samples can be dissolved in ddH2O or TE buffer (pH 8.0).
Whole Genome Sequencing (Nanopore)
Through the third generation sequencing platform Nanopore, genome sequencing of individuals with known or unknown DNA sequences is conducted. The characteristic of long-read data in Nanopore sequencing provides better results in genome assembly, identification of structural variations, and recognition of continuous repeat sequences. This feature leads to a broader range of applications in areas such as genomics research, biomedical sciences, and biotechnology.
Nanopore sequencing can be paired with chips of different specifications for selection. The report includes quality control (QC) of samples and data, assembly and alignment of sequences (BlastN, BlastX), database analysis (GO, COG, KEGG), and Genome map, as well as customized analysis services.
Sample Requirements :
Sample Purity: OD 260/280 ratio should be between 1.7 and 1.9.
Sample Concentration: Minimum concentration should not be less than 100 ng/µl.
Total Sample Amount: The recommended total amount is 5 µg, with a minimum of at least 1 µg.
Sample Solvent: Samples can be dissolved in ddH2O or TE buffer (pH 8.0).

Whole Exome Sequencing
Whole Exome Sequencing (WES)
Whole Exome Sequencing (WES) is a high-throughput genomic technique that differs from Whole Genome Sequencing (WGS) in its focus on sequencing the exonic regions of an organism's genome. Exons are the regions of the genome that encode proteins and are generally considered the most functionally important gene regions in an organism. Despite the human genome containing approximately 3 billion base pairs, exons make up only about 1% of the total genome. However, they harbor the majority of mutations responsible for diseases. Therefore, Whole Exome Sequencing is highly valuable for identifying variations in genes that encode proteins, contributing significantly to the understanding of genetic diseases and personalized medicine. The main features of Whole Exome Sequencing (WES) include:
Targeted Sequencing: Unlike sequencing the entire genome, WES selectively focuses on the exonic regions of genes. Exons are DNA segments transcribed into messenger RNA (mRNA), ultimately used in protein synthesis.
Cost-Effective: Compared to Whole Genome Sequencing (WGS), WES is more cost-effective as it sequences only a small portion of the genome while providing crucial information about protein-coding regions. This is particularly useful in many research and clinical applications.
Variant Detection: WES is primarily used for detecting single nucleotide variations (SNVs) and small insertions or deletions (indels) within exons. These variations may include mutations causing diseases as well as common genetic polymorphisms.
Clinical Applications: WES plays a crucial role in identifying rare Mendelian genetic diseases, cancer genomics, and understanding the genetic basis of different diseases. It aids in diagnosing and predicting the disease risk for patients.
Cancer Genomics: In cancer research, WES is often employed to identify somatic mutations in tumor genomes, contributing to treatment decisions and the development of targeted therapeutic approaches.
Sample Requirements :
Sample Purity: The OD 260/280 value should be between 1.7 and 1.9. RNA and protein contamination should be thoroughly removed.
Sample Amount: A recommended minimum of 1 µg total sample is advised (at least 10 ng).
Sample Solvent: Samples can be dissolved in H2O or TE (pH 8.0). Alternatively, samples can be shipped in a dry state.
16S/ITS Metagenomics
16S/ITS Metagenomics
Metagenomics, also known as Environmental Genomics, is the scientific study of an entire microbial community. It involves extracting DNA from environmental samples containing microorganisms, sequencing the DNA, and then conducting analyses such as species identification, phylogenetic assessments, gene function studies, and phenotype correlations. The aim is to gain insights into the diversity of microorganisms present in the environment.
The 16S rRNA gene is present in the ribosomes of all bacteria and exhibits highly conserved sequences as well as diverse features among different bacterial species. Consequently, it serves as a valuable marker for bacterial classification and identification. 16S metagenomics primarily focuses on the sequencing of the 16S rRNA gene sequences of bacteria. This involves amplifying rRNA fragments through PCR, constructing paired-end libraries, and utilizing the Illumina MiSeq platform for sequencing. Subsequently, the obtained sequences are analyzed to determine the bacterial taxa and their distribution proportions in each sample based on sequence variations and abundance.
In summary, the application of metagenomics provides a comprehensive and in-depth understanding of microbial community structure and function. This approach is crucial for ecological studies, environmental science, and research related to microbial health.
Sample Requirements :
Sample Purity: The OD 260/280 ratio should fall within the range of 1.7 to 1.9.
Total Sample Amount: It is recommended that the total amount of each sample is not less than 50 ng.
Sample Solvent: Samples can be dissolved in ddH2O or TE buffer (pH 8.0).
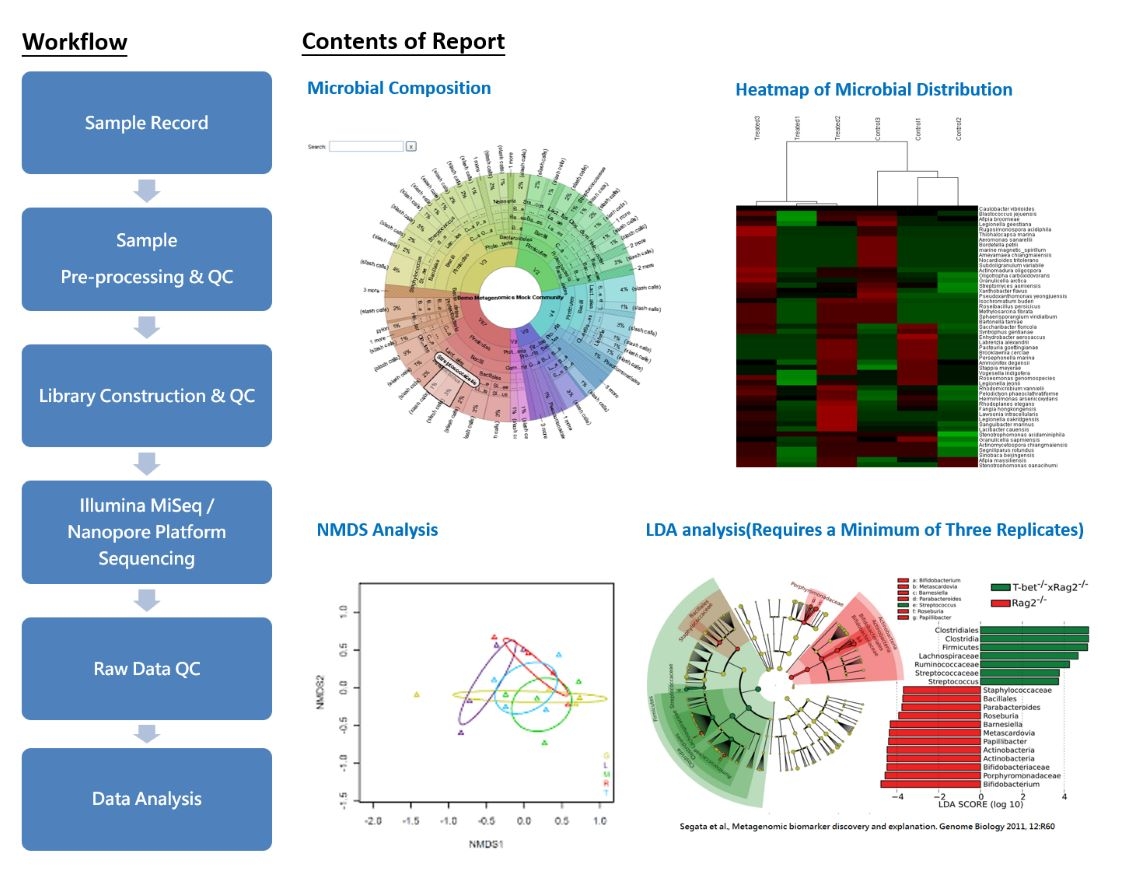
Whole Genome Metagenomics
Whole Genome Metagenomics (Shotgun Metagenomics)
Metagenomics, also known as Environmental Genomics, is the scientific study of an entire microbial community. In this field, DNA is extracted from environmental samples containing microorganisms, sequenced, and then subjected to analyses such as species identification, phylogenetics, gene function, and phenotype correlations. The goal is to gain insights into the diversity of microorganisms present in the environment.
Whole genome metagenomics represents a more comprehensive research approach that enables the acquisition of genomic sequence information for all microorganisms within a specific environmental microbial community. By overcoming the limitations of microbial isolation and cultivation techniques, this method directly extracts DNA from environmental samples for sequencing. Through high-throughput DNA sequence analysis, the collective genomic information for each microorganism is obtained. This information is then utilized to study microbial community aspects such as taxonomic composition, distribution proportions, phylogenetic relationships, gene functions, and metabolic pathways. By providing deeper genetic insights, researchers can gain a more comprehensive understanding of the ecology and metabolic functions of microbial communities.
Sample Requirements :
Sample Purity: OD 260/280 ratio should be between 1.7 and 1.9.
Sample Concentration: Minimum concentration should not be less than 30 ng/µl.
Total Sample Amount: The recommended total amount is 5 µg, with a minimum of at least 1.5 µg.
Sample Solvent: Samples can be dissolved in ddH2O or TE buffer (pH 8.0).


RAD-seq (ezRAD-seq)
Restriction-site Associated DNA Sequence (RAD-seq) (ezRAD-seq)
RAD-seq (Restriction-site Associated DNA Sequence) refers to the use of restriction enzymes to cleave the genome, significantly reducing its complexity. This technique involves high-throughput sequencing of specific-length enzyme-digested fragments (RAD-tags) to rapidly identify highly accurate Single Nucleotide Polymorphisms (SNPs), facilitating analyses such as population evolution, genetic map construction, and QTL (Quantitative Trait Locus) localization.
On the other hand, ezRAD-seq is an improved version of RAD-seq technology that simplifies experimental procedures by utilizing sequencing library kits. This allows for the rapid adoption of the technique in general laboratories without the need for extensive experimental equipment or complex steps, enabling the generation of a large amount of SNP data while minimizing time and experimental costs.
Furthermore, ezRAD-seq, based on SNP molecular marker technology, offers a high cost-performance ratio and excellent stability, making it particularly suitable for the analysis of a large number of samples. Currently, this technique is widely applied in the study of genetic diversity in non-model organism, molecular breeding, systematics, and population genomics, among other fields.
Sample Requirements :
Sample Purity: The OD 260/280 ratio should be between 1.7 and 1.9.
Sample Total Amount: It is recommended that each sample has a total amount of no less than 2 μg DNA.
Sample Solvent: Samples can be dissolved in ddH2O or TE buffer (pH 8.0).

ChIP-seq
Chromatin Immunoprecipitation Sequencing (ChIP-Seq)
Chromatin Immunoprecipitation (ChIP) is a crucial technique for studying the interactions between DNA and proteins within the cell. It sensitively detects the binding of specific proteins to particular DNA segments. As eukaryotic genomic DNA exists in a chromatin form, interactions between DNA and proteins within the chromatin elucidate the fundamental pathways of gene expression in eukaryotes. Through the ChIP technique, researchers gain insights into the interactions between chromatin and its associated regulatory factors, typically employed to study transcription factor binding sites or sites of histone-specific modifications. Chromatin Immunoprecipitation Sequencing (ChIP-Seq) merges ChIP with Next Generation Sequencing (NGS) technology. It involves high-throughput sequencing of obtained DNA fragments, generating millions of sequences precisely mapped onto the genome, aiming to extract biological information about DNA segments involved in interactions with proteins like histones or transcription factors.
Sample Requirements :
Sample Purity: O.D. 260/280 ratio should be between 1.7 to 1.9; Ensure removal of RNA and proteins.
Sample Concentration: Minimum concentration should not be lower than 5 ng/µl (If the sample requires QC, concentration should not be lower than 20 ng/µl).
Sample Total Amount: Each sample should have a total amount not less than 10 ng (If the sample requires QC, total amount should not be lower than 100 ng).
Sample Solvent: Samples can be dissolved in ddH2O or TE buffer (pH 8.0).

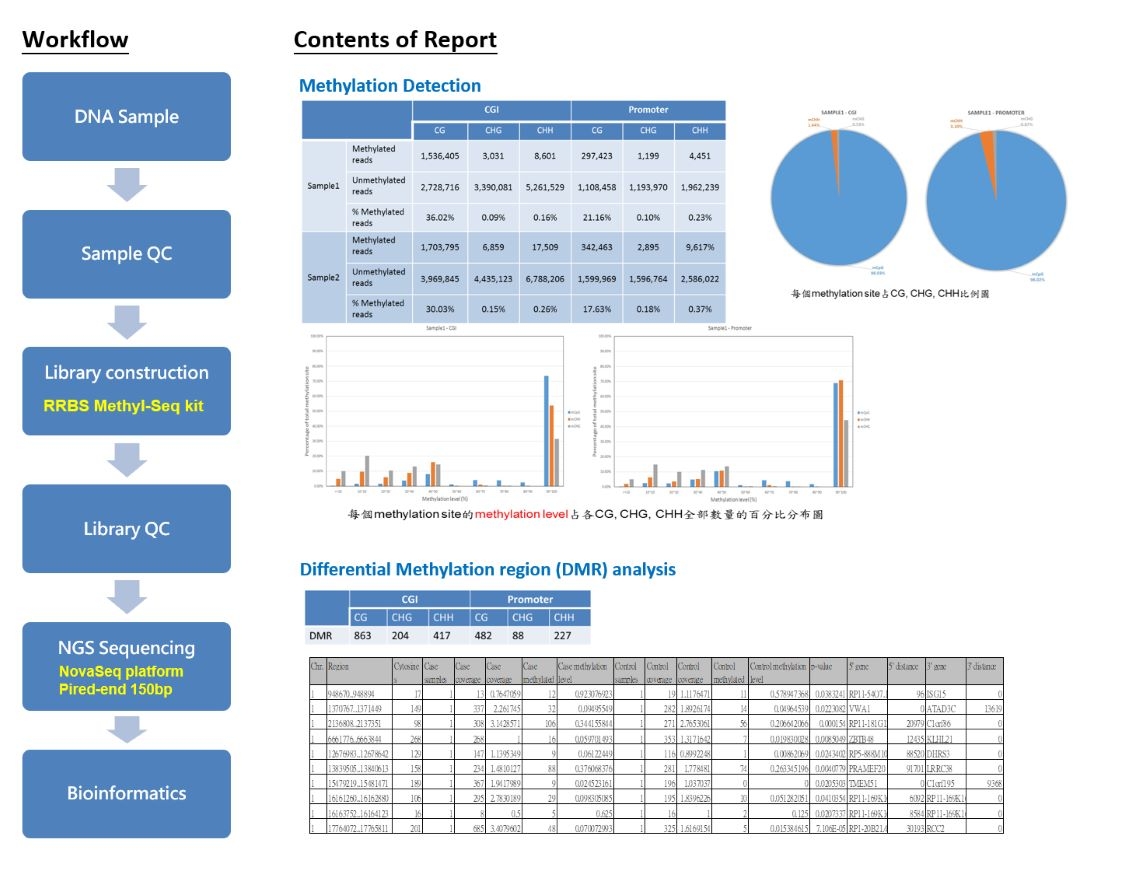
RRBS-seq
Reduced Representation Bisulfite Sequencing (RRBS-Seq)
Reduced Representation Bisulfite Sequencing (RRBS) is a precise, efficient, and cost-effective method for studying DNA methylation. It involves enzymatic enrichment of promoter and CpG island regions, followed by bisulfite sequencing, enabling high-resolution detection of DNA methylation status with high utilization of sequencing data. DNA methylation research has long been a focal point in disease studies, closely associated with gene expression and phenotypic traits. RRBS, as a cost-effective methylation analysis method, holds extensive potential for research in large-scale clinical samples.
Sample Requirements :
Sample Purity: O.D 260/280 ratio should be between 1.7 and 1.9; RNA and protein should be thoroughly removed.
Sample Concentration: Minimum concentration should not be lower than 50 ng/µl.
Sample Total Amount: At least 1 µg per sample (recommended 3 µg).
Sample Solvent: Samples can be dissolved in ddH20 or TE buffer (pH 8.0).
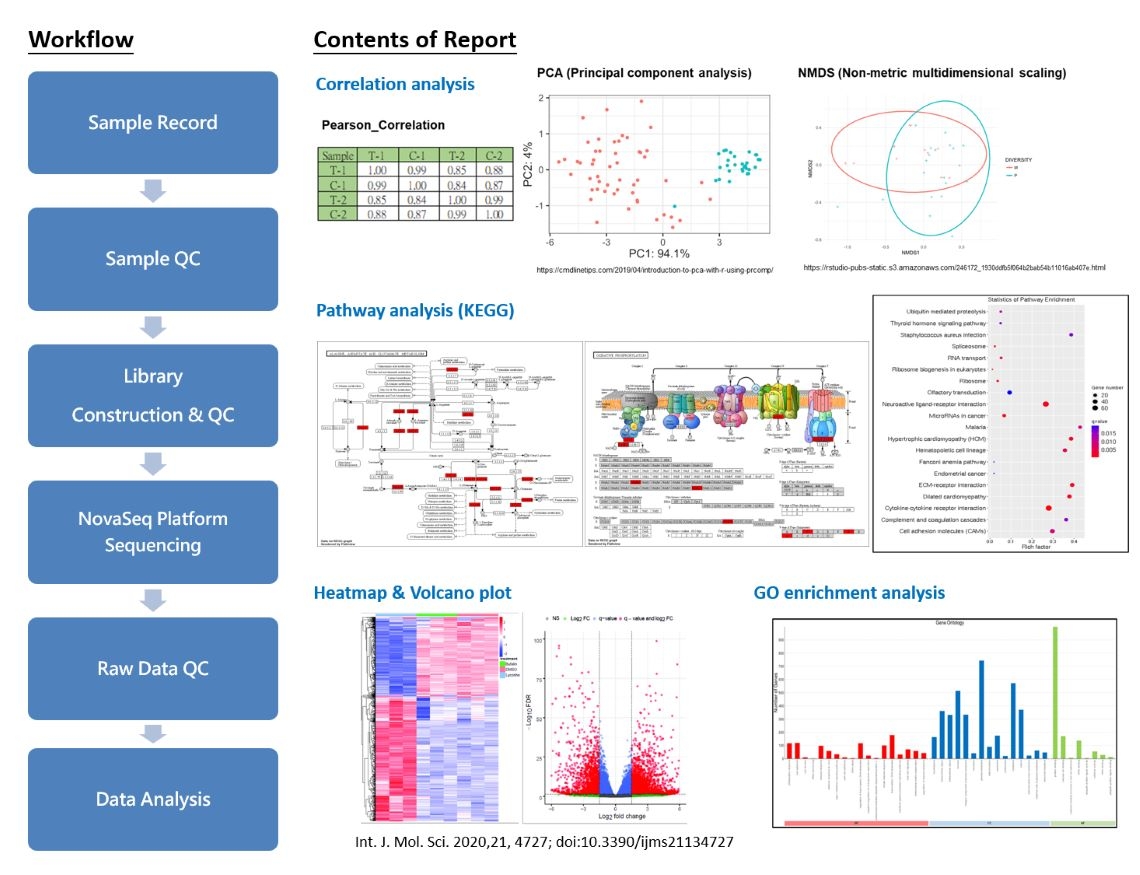
RNA-Seq
RNA Sequencing (Transcriptome)
RNA sequencing, commonly abbreviated as RNA-seq, is a powerful and widely used technique in molecular biology and genomics for analyzing the transcriptome of cells or tissues. The transcriptome represents the complete set of RNA molecules produced by a specific cell or tissue at a particular time, including messenger RNA (mRNA), non-coding RNA, and other functional RNA.
As library construction and sequencing involve a "random" sampling process, it cannot guarantee the successful sequencing of every fragment. Additionally, genes exhibit varying expression levels, with highly expressed genes having a greater abundance of RNA fragments, increasing the probability of being sampled during sequencing. Conversely, lowly expressed genes have fewer RNA fragments, resulting in a relatively lower chance of being sampled. With the advancement of next generation sequencing (NGS), which provides high-throughput gene sequencing, RNA-seq has become a robust tool for transcriptomic studies.
The development of NGS technology allows the fragmentation of RNA molecules of varying lengths into 150 bp fragments. Through PCR amplification, this approach enables the generation of extensive data, covering the complete RNA information and providing accurate quantitative data.
RNA-seq finds applications in various analyses, including gene expression analysis, alternative splicing analysis, and exploration of non-coding RNAs such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs). Recently, the integration of RNA-seq with single-cell technologies has emerged, allowing the analysis of the transcriptome at the single-cell level and facilitating research into cellular heterogeneity within tissues.
Sample Requirements:
Sample Purity: The OD 260/280 value should be between 1.9 and 2.1. DNA and protein contamination should be thoroughly removed.
Sample Amount: A recommended minimum of 1 µg total sample is advised (at least 10 ng).
Sample Solvent: Samples can be dissolved in H2O or TE (pH 8.0). Alternatively, samples can be shipped in a dry state.
Small RNA seq
Small RNA Sequencing (microRNA Sequencing)
MicroRNAs (miRNAs) are endogenous, single-stranded, non-coding RNA molecules, approximately 22 nucleotides in length. They play a crucial role in post-transcriptional gene silencing pathways, suppressing protein synthesis and influencing the expression of proteins involved in cell development, proliferation, differentiation, and apoptosis.
Since their discovery in 1993, miRBase has annotated over 28,645 miRNAs, estimated to regulate around 30% of the human genome. Each miRNA can regulate hundreds of genes, linking them to cell differentiation, proliferation, and programmed cell death. Consequently, they affect systems such as stem cells, immunity, and cancer. With the increasing research in oncology, variations in miRNA expression have been found in different cancers. They can regulate genes involved in tumor initiation and progression, making them useful for cancer classification, diagnosis, prognosis, and potential therapeutic developments.
Sample Requirements :
Sample Purity: OD 260/280 ratio should be above 1.8; DNA and proteins should be thoroughly removed.
Sample Concentration: Total RNA concentration should not be lower than 25 ng/µl (2 ng/µl is acceptable for Risk library construction).
Sample Amount: Each sample should have a total amount of at least 1000 ng (2 ng/µl is acceptable for Risk library construction).
Sample Solvent: Samples can be dissolved in H2O or TE (pH 8.0) or sent in a dried state.

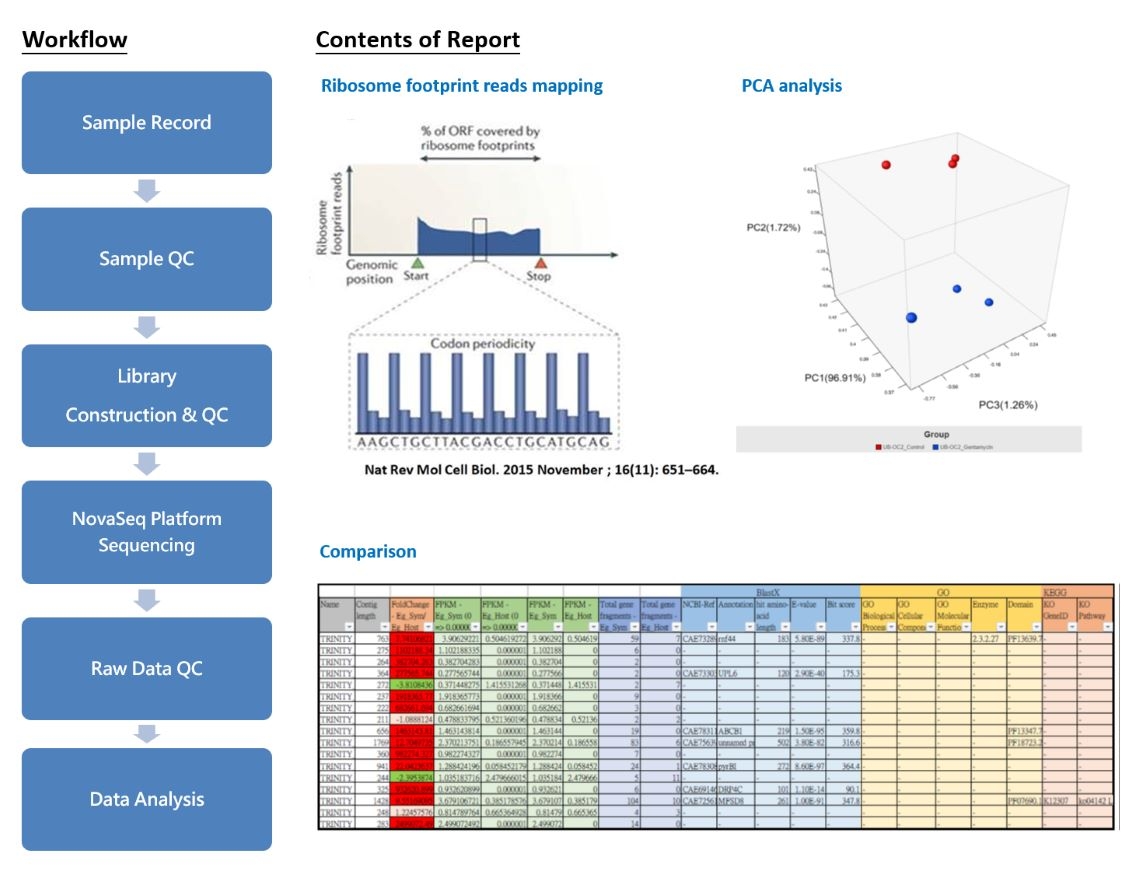
Ribo-seq
Ribo-seq (Ribosome profiling)
Ribo-seq, also known as Ribosome profiling, is a novel technology developed based on RNA-seq. The main difference lies in the process of handling samples in Ribo-seq, where RNase is introduced to cleave RNA without ribosome protection. This method specifically analyzes RNA fragments that are actively undergoing translation and bound to ribosomes, known as ribosome-protected RNA fragments (RPFs). This approach allows for a deeper understanding of translational regulation mechanisms.
As a crucial tool for deciphering protein synthesis regulation, our service employs the RiboLace technology developed by IMMAGINA BioTechnology. This technology utilizes magnetic beads for affinity purification of translating ribosomes, followed by the collection of RPFs for high-throughput sequencing. In comparison to traditional Ribo-Seq, our method requires only a small amount of sample (>0.3M cells), eliminates the need for gel purification steps, and streamlines the experimental timeline from 10 days to 2~3 days. This not only significantly lowers the sample requirement threshold but also accelerates the data acquisition process.
Sample Requirements :
Sample Purity: The OD 260/280 ratio should be between 1.7 and 1.9.
Sample Total Amount: It is recommended that each sample has a total amount of no less than 1.5 μg RNA.
Sample Concentration: The concentration should be above 25 ng/μl (Bioanalyzer quality testing requires a concentration above 25 ng/μl).
Sample Solvent: Samples can be dissolved in ddH2O or TE buffer (pH 8.0).

Single-cell RNA-seq
Single cell RNA-seq (scRNA-seq)
Single-cell RNA sequencing (scRNA-seq) is an emerging and powerful molecular biology technique used to analyze gene expression at the level of individual cells. While traditional RNA sequencing provides information on the average gene expression across a large number of cells, scRNA-seq provides data on the gene expression of each individual cell. This enables researchers to examine the gene expression profiles of individual cells within a heterogeneous population, allowing deeper insights into cellular diversity, differentiation, and other biological processes. As a result, scRNA-seq has broad applications in developmental biology, immunology, neuroscience, regenerative medicine, and drug discovery.
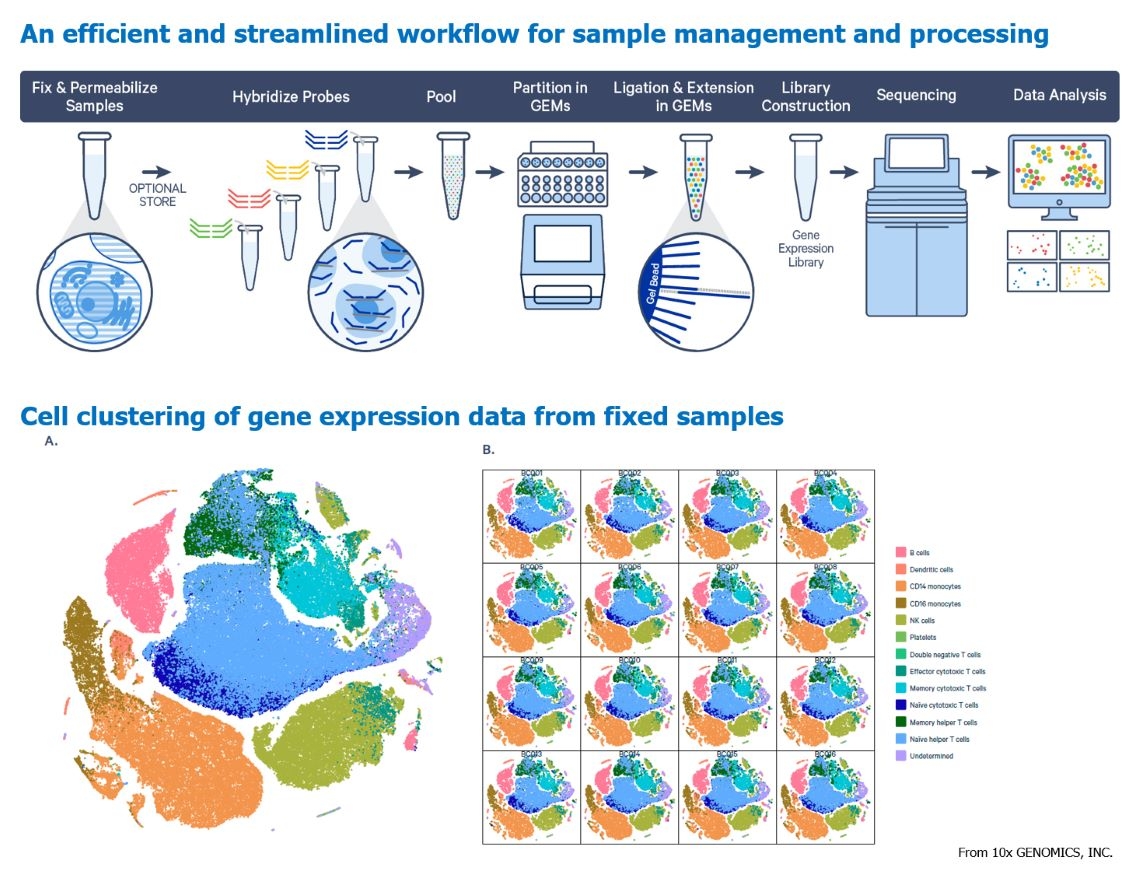
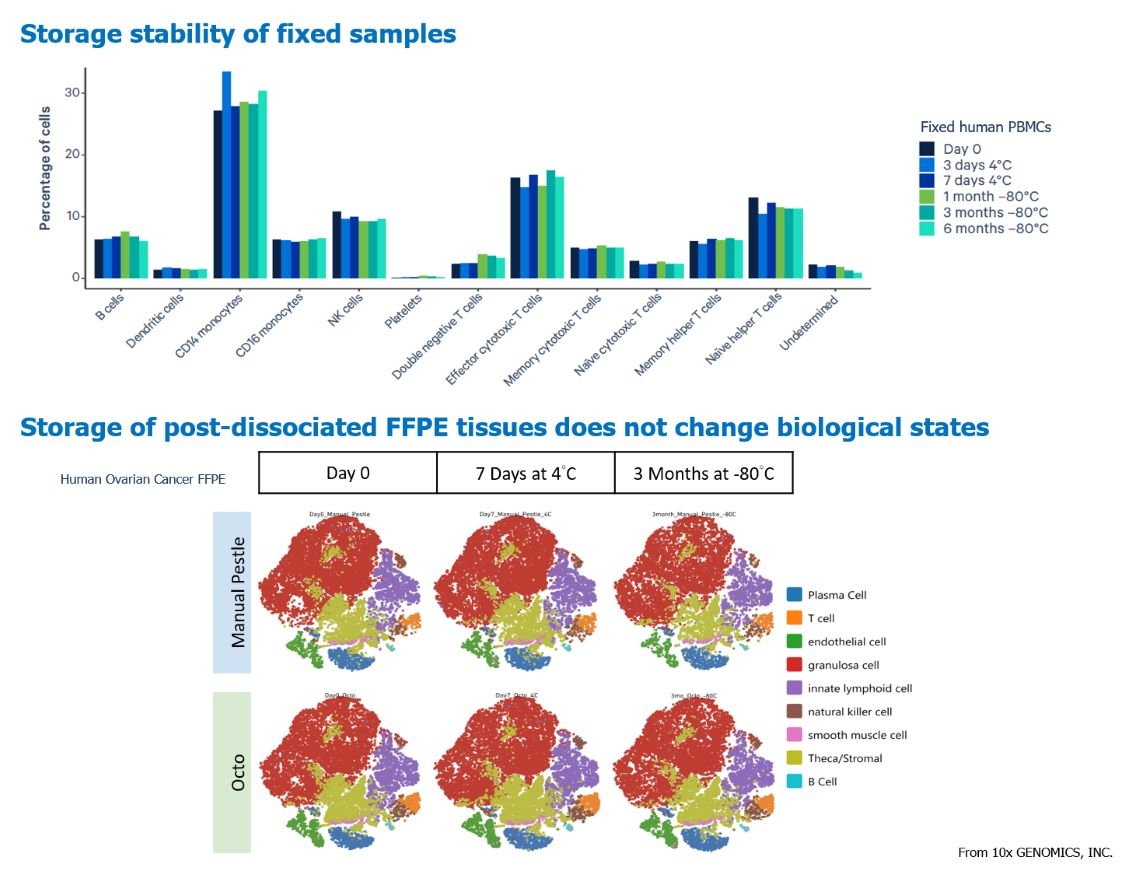
Recently, 10x GENOMICS, INC has introduced new assay kits that use Fixation Buffer to preserve live cells, thereby locking in the RNA expression state of each cell. Subsequent reactions with probes designed to detect gene expression enable the measurement of gene expression levels in each cell. Cells that have been fixed can be stored at -80℃ for up to six months while retaining their original gene expression profiles. This approach can even be applied to FFPE (formalin-fixed paraffin-embedded) specimens, reducing the need for repeated experiments using only live cells for single-cell sequencing. This significantly decreases time pressure, batch-to-batch variations, and minimizes reagent consumption costs, maximizing the potential of scRNA-seq on a larger scale.
Strain Identification
Advantages of Strain Identification Service :
1. Genetic identification methods analyze bacterial 16S and fungal ITS, capable of identifying at the "species" level.
2. Both agar plates and liquid cultures of individual strains are applicable. Genomic DNA of microorganisms can also be submitted for identification.
3. Reports are generated within 5 working days upon sample receipt.
4. A dedicated data management system is available for customers to download files.